汞是唯一以气态单质形式在大气中远距离迁移的重金属。大气中氧化还原过程能改变汞的化学形态,影响汞的环境行为和生态效应。尤其重要的事,汞的氧化会直接导致惰性气态单质汞转变为高活性气态氧化汞(GOM)。后者极易吸附在颗粒物上形成颗粒态汞(PBM),与GOM会通过干湿沉降进入地表。其中部分可转化为剧毒甲基汞,对生态环境造成严重危害。
关于大气汞的主要氧化途径(Br∙、Cl∙、OH∙、O3等),仍存在诸多争议。稳定同位素为探究大气汞的氧化提供关键证据。近日,地球系统科学学院同位素前沿科学研究中心陈玖斌教授等联合国内外多家科研单位,对从雪龙号上收集到的往返上海和南极的海相气溶胶Hg和S同位素进行了系统研究,发现跨南北半球海洋气溶胶中Hg和S同位素组成随纬度协同变化,判定其主要原因是从赤道到极地大气Hg0氧化剂明显变化,从热带地区以OH∙、O3氧化为主转变到极地地区以Br∙、Cl∙为主,并提出PBM产生的特殊氧化过程。该研究为量化全球大气Hg、S甚至N通量、评估大气颗粒物对气候和生态系统的响应和影响提供了新思路。
发现:汞、硫同位素非质量分馏的纬度变化
汞具有三维同位素示踪体系,分别是质量分馏(MDF)、奇数和偶数非质量分馏(odd-MIF和even-MIF)。北半球亚热带(0°-30°N)和南半球(0°-70°S)颗粒物样品的Δ199Hg、Δ200Hg和Δ33S值存在明显空间差异行,表现为南半球样品的同位素组成与纬度呈显著正相关(图1),而北半球亚热带样品的同位素组成未见明显纬度变化,这很可能反映了不同的Hg氧化途径。此外,数据显示,第一个航程(08/11/15至25/11/15)颗粒物样品的Δ33S与Δ200Hg显著相关,而与Δ199Hg不相关;第二个航程(26/11/15至10/04/16)颗粒物样品Δ33S与Δ199Hg显著相关,而与Δ200Hg不相关。上述结果说明采样期间至少涉及两类反应(或MIF生成机制):一种造成南半球Hg同位素(如Δ199Hg、Δ201Hg和Δ200Hg)和S同位素(Δ33S < 0.20‰)随纬度梯度变化,另一种导致样品Δ33S和Δ200Hg同步正偏。
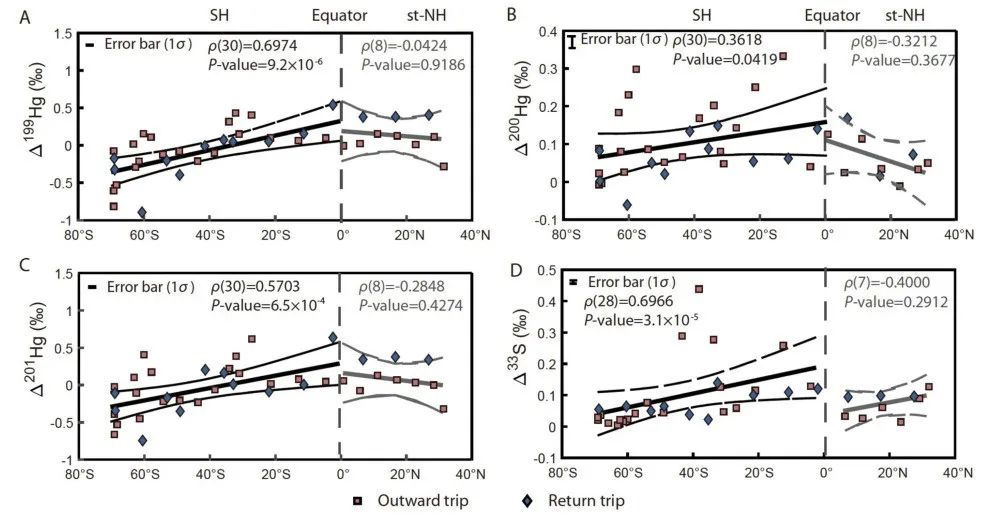
图1:巡航往返期间从北半球亚热带到南半球颗粒物样品Hg、S同位素随纬度变化图
氧化剂转变引起汞、硫同位素非质量分馏的纬度变化
如图2,在连续4个月的往返航程中观察到的对称模式表明,南半球颗粒物样品出现奇数Hg同位素非质量分馏(odd-MIF)和Δ33S的正纬度梯度变化是常年特征,很可能受海洋边界层中O3、OH∙、卤素化合物(Br∙和Cl∙)等氧化剂变化的控制。该研究使用大气化学传输模型(GEOS-Chem)评估了四种氧化剂OH∙、O3、Br∙、Cl∙对海洋边界层和对流层底部Hg0氧化的年度空间贡献,结果显示,在0°-40°S,以OH∙和O3为主的Hg0氧化约占80%,而40°S-80°S以Br∙为主的氧化约占75%。因此,不同氧化途径可能是造成汞同位素组成变化的根本原因。
对于S体系,南半球参与生源硫化物二甲基硫(DMS)氧化的气相 BrO、OH∙、O3也呈现出空间分布变化,在0°-40°S,OH∙/O3主导的氧化占60%,而在40°S-80°S,BrO主导的氧化占70%,DMS主要氧化剂由O3/OH∙到卤素化合物的转变与GEOS-Chem模拟结果一致。因此,氧化途径的转变是导致汞、硫同位素协同变化的主因。
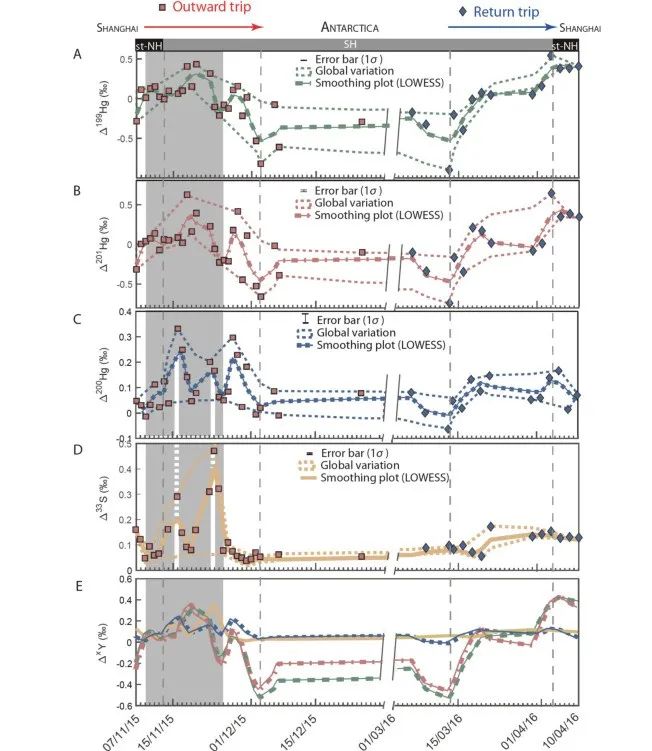
图2:颗粒物样品Hg、S同位素随采样时间变化图
矿物粉尘上的非均相光反应可能触发偶数汞同位素异常
过去普遍认为Δ200Hg和Δ33S与平流层硫酸盐的输入有关。但样品Δ33S最高为0.44‰,远远低于平流层硫酸盐Δ33S(最大可达10‰)。而气团轨迹模型也表明,对流层上层或平流层Hg和S的输入不是造成Δ200Hg和Δ33S偏移的原因。
最近研究表明,矿物粉尘存在下SO2的光氧化会产生明显33S异常,这很可能解释上海和澳大利亚往返航程间的Δ33S差异。与其他季节不同,澳大利亚9-12月经常发生沙尘暴。采样期间的气团轨迹模型(HYSPLIT)模拟的沙尘粒子前向轨迹也证明了这种可能性。样品中测得的Δ33S和Δ36S分别高达0.44‰和1.21‰,目前只能通过光氧化来解释。因此,同步变化的Δ200Hg正偏(高达0.30‰)很可能也是矿物粉尘表面Hg0的光催化氧化过程造成。
南极洲大气汞相态转变
研究观察到,在南极停留期间采集的样品Δ 199 Hg和Δ 201 Hg值偏负(范围为-0.50‰至-0.30‰),且Hg浓度骤然增加。这一现象可能与南极汞连续的氧化还原反应相关。极地地区春季海冰和积雪会释放大量Br,会导致Hg 0 氧化和Hg II 沉降,即大气汞损耗事件(AMDE)。随后大量沉降在积雪中的Hg II 经光还原反应重新释放回大气,再被重新氧化,最终在大气颗粒物上富集。这一基于同位素数据所得出的结论,被南极春夏季汞循环模型中的氧化还原过程的模拟结果和实地观测数据所验证。
启示:对大气氧化的研究意义
跨半球巡航(30°N-60°S)收集的气溶胶中Hg-MIF的纬度变化表明,由Br∙引发的两步氧化不是南半球海洋边界层Hg0氧化的唯一机制。S和Hg(也包括N)的模拟氧化也表明,主要氧化剂从南半球亚热带的OH∙和O3,转变为中纬度和极地的Br∙。气溶胶中S、O和Hg在亚热带地区显示出更高的MIF,这与该地区OH∙、O3对Hg0氧化更强有关。以前的全球汞模型研究将OH∙、O3和Br单独作为模型中汞的主要氧化剂,但该研究表明,两种氧化途径都在海洋边界层的不同区域共同存在,应综合考虑。
同时,该研究指出S-MIF和Hg-MIF可能受澳大利亚大陆排放的矿物粉尘上的非均相光反应控制。因此,气溶胶表面的非均相氧化可能补充全球大气汞模型中氧化过程的缺失。
此外,该研究揭示的氧化途径在高O3区域可能普遍存在,很可能揭示城市雾霾过程中PBM和PM2.5浓度的正相关关系。
最后,上述氧化途径会减少Hg0在大气中的滞留时间,增加区域HgII沉降速率,不但改变HgII和Hg0通传输量,也会影响区域Hg的生态环境效应。
David AuYang, Jiubin Chen, Wang Zheng, et al. South-hemispheric marine aerosol Hg and S isotope compositions reveal different oxidation pathways. National Science Open 2022; 1: 20220014
https://doi.org/10.1360/nso/20220014

